Chromatography System Qualification
1. Purpose and Scope
Chromatography systems used in GMP manufacturing are direct-impact purification equipment. They directly influence product purity, impurity clearance, yield, and batch consistency. Qualification must demonstrate that the integrated system performs as intended under defined operating conditions and remains in a validated state throughout its lifecycle.
This article addresses qualification of:
- Chromatography skid hardware
- Column integration and mechanical integrity
- Instrumentation and control systems
- Software and data integrity controls
- Cleaning and sanitization strategy
- Single-use system considerations
- Requalification and continued verification
The diagram below defines the qualification boundary of a chromatography system within a GMP environment. It identifies the direct-impact components requiring structured qualification, including mechanical hardware, control systems, instrumentation, column integration, and cleaning or disposable flow paths. Establishing clear system boundaries prevents scope ambiguity and ensures that risk assessment and qualification activities address all product-contact and control-critical elements.
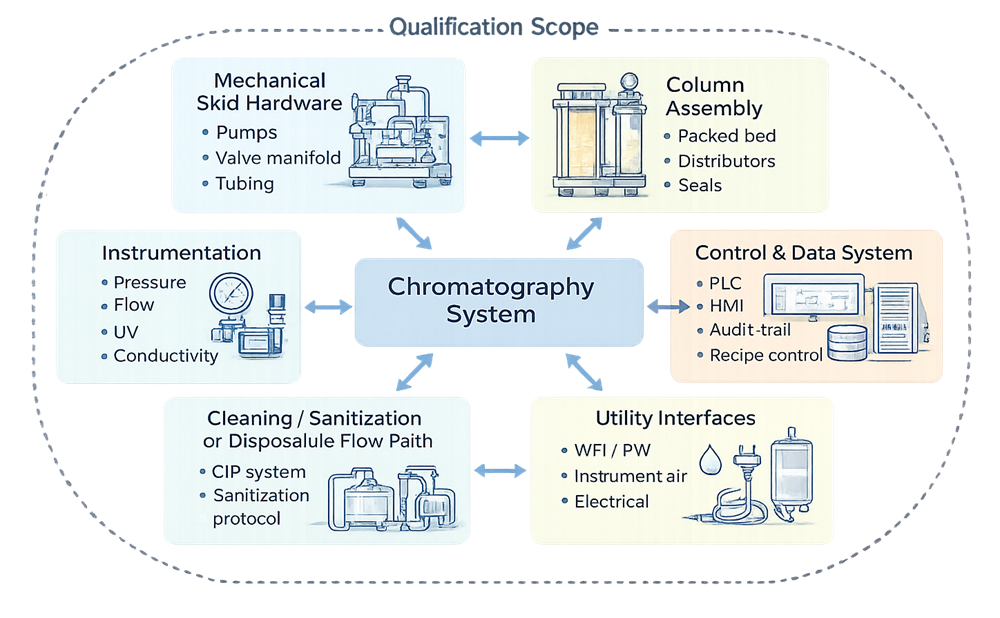
Process performance validation of the purification step is addressed separately within process validation programs. This article focuses on equipment and system qualification.
2. Qualification Lifecycle Strategy
Chromatography systems follow a structured lifecycle model:
• Design Qualification
• Installation Qualification
• Operational Qualification
• Performance Qualification
• Continued Process Verification
The depth of qualification is determined by:
• System complexity
• Automation level
• Cleaning strategy
• Reuse model
• Risk assessment outcome
Reusable stainless steel systems require broader qualification scope than single-use platforms.
3. Design Qualification
Design Qualification confirms that the selected system meets the User Requirement Specification and is suitable for its intended purification application. DQ should verify:
• Pressure and flow range compatibility with process requirements
• Wetted material chemical compatibility
• Surface finish for stainless steel components
• Valve configuration and flow path logic
• Instrument accuracy and range
• Control system architecture
• Data management capabilities
For automated systems, DQ must confirm:
• Role-based access control
• Audit trail capability
• Electronic record retention
• Alarm configuration logic
DQ establishes traceability between user requirements, risk assessment, and system design.
4. Installation Qualification
Installation Qualification verifies that the system is installed according to approved design documentation and manufacturer specifications.
IQ typically includes:
• Equipment identification and asset tagging
• Verification of materials of construction
• Utility connection confirmation
• Calibration status of instrumentation
• Wiring and cabinet inspection
• Software version documentation
• Spare parts verification
For stainless steel systems:
• Weld documentation review
• Surface finish confirmation
• Drainability assessment
• Gasket and seal verification
For single-use systems:
• Frame integrity
• Pump installation verification
• Control configuration
• Disposable component compatibility confirmation
IQ establishes documented installation compliance.
5. Operational Qualification
Operational Qualification demonstrates that the chromatography system operates within predefined limits and that control logic functions correctly. OQ typically challenges:
• Flow rate accuracy and repeatability
• Pressure monitoring and interlocks
• Valve sequencing
• Fraction collection switching logic
• Sensor accuracy and alarm functionality
• Recipe execution and parameter control
Software verification includes:
• User access level testing
• Audit trail verification
• Data integrity checks
• Electronic record generation
OQ confirms equipment functionality independent of product-specific validation.
6. Performance Qualification
Performance Qualification demonstrates that the integrated system performs reproducibly under actual or simulated production conditions. PQ typically evaluates:
• Cycle-to-cycle reproducibility
• Pressure stability
• Peak profile consistency
• Binding capacity performance
• Resolution and asymmetry stability
• Fraction collection accuracy
For reusable columns, PQ may incorporate:
• Multi-cycle performance trending
• Regeneration effectiveness
• Sanitization impact assessment
PQ confirms that the system supports validated purification processes.
7. Cleaning and Sanitization Qualification
Cleaning and sanitization qualification must demonstrate that product-contact surfaces are returned to a defined state of cleanliness and microbiological control without compromising column integrity or resin performance.
The following illustration differentiates cleaning validation from sanitization qualification within chromatography systems. Cleaning focuses on removal of chemical and product residues to predefined acceptance limits, while sanitization addresses microbiological control and resin compatibility. The diagram clarifies how these two control strategies operate under distinct validation objectives.
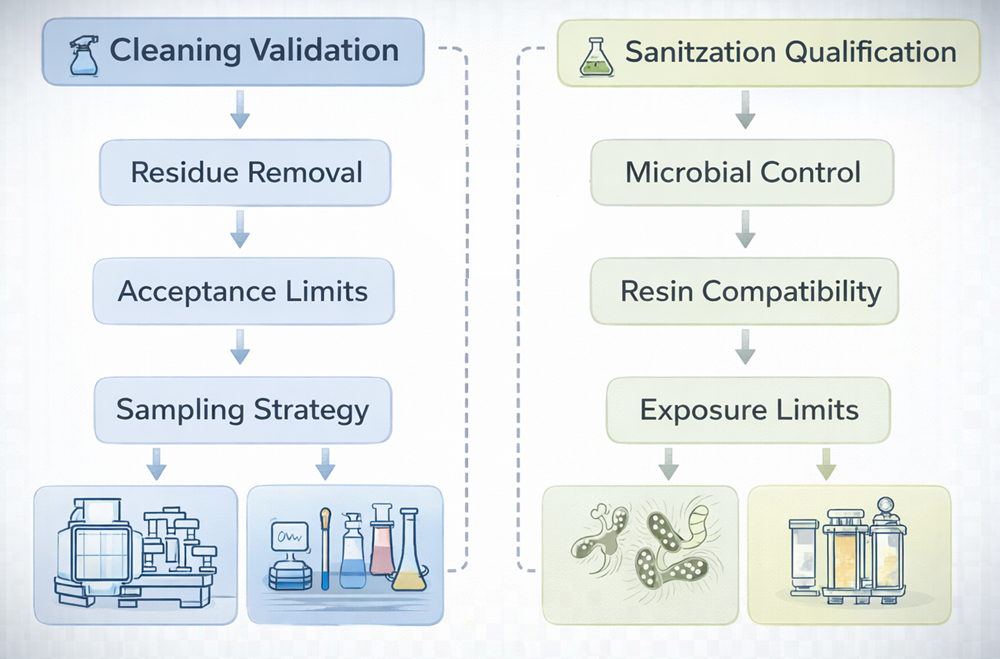
7.1 Cleaning Validation – Stainless Steel Systems
Cleaning validation applies to:
• Column hardware and distributors
• Valve manifolds
• Pumps and tubing
• Fraction collection lines
Cleaning must effectively remove:
• Product residues
• Process impurities
• Cleaning agents
• Endotoxins where applicable
Acceptance limits must be scientifically justified based on:
• Health-based exposure limits
• Toxicological data
• Maximum allowable carryover calculations
Verification methods may include:
• Swab sampling
• Rinse sampling
• TOC analysis
• Specific analytical assays
Worst-case conditions must address:
• Maximum campaign length
• Worst-case residue
• Dirty hold time
• Clean hold time
Reproducibility must be demonstrated across multiple validation runs.
7.2 Sanitization Qualification
Sanitization is focused on microbiological control. Qualification must demonstrate:
• Defined bioburden reduction
• Effective sanitant distribution throughout the bed
• No unacceptable degradation of ligand functionality
Common sanitization agents include sodium hydroxide at validated concentration and contact time. Impact assessment must confirm no unacceptable decline in:
• Binding capacity
• Backpressure
• Peak symmetry
• Resolution
Cumulative chemical exposure limits should be defined.
7.3 Single-Use System Qualification
Single-use systems eliminate traditional cleaning validation but require alternative controls. Qualification must address:
• Supplier qualification
• Component lot traceability
• Extractables and leachables risk assessment
• Pre-use integrity testing
• Compatibility with process buffers
Integrity verification may include pressure hold testing or visual inspection. Disposable flow paths shift risk control from cleaning validation to supplier management and component verification.
The illustration below contrasts qualification scope between stainless steel and single-use chromatography systems. Reusable stainless steel platforms require cleaning validation, sanitization qualification, and reuse justification, whereas single-use systems shift focus to supplier qualification, material traceability, extractables and leachables assessment, and pre-use integrity verification.
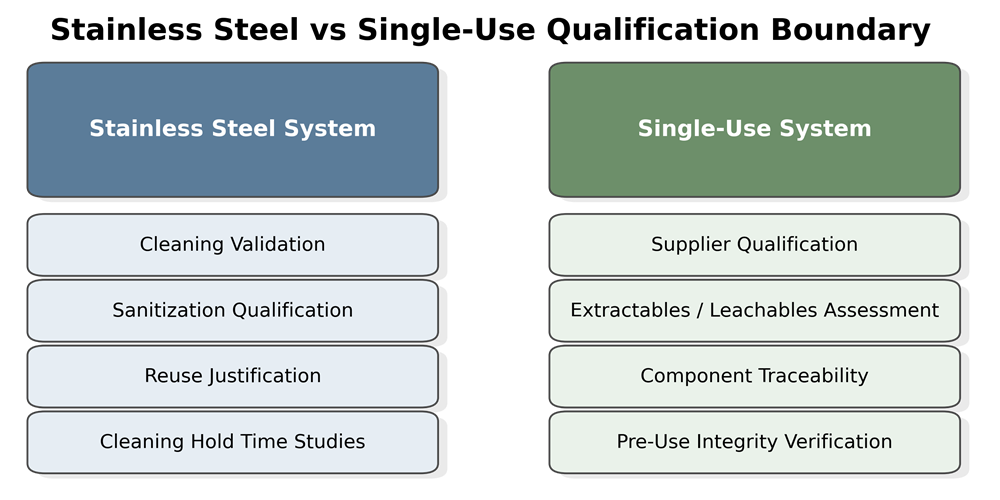
8. Instrumentation and Data Integrity
Instrumentation qualification must confirm:
• Calibration traceability
• Accuracy across operating range
• Alarm setpoint verification
Control systems must ensure:
• Secure user access
• Audit trail integrity
• Version control
• Change management
Systems generating electronic records must comply with applicable regulatory requirements.
9. Requalification and Continued Verification
Chromatography systems operate within a controlled lifecycle framework. Requalification is not automatic for every change; it is risk-based and proportional to impact on product-contact surfaces, validated operating parameters, and system performance. Requalification may be required following:
• Major component replacement
• Control system modification
• Cleaning or sanitization process change
• Column type or pressure rating change
• Repeated or unexplained performance deviations
The extent of requalification must be justified through documented impact assessment.
9.1 Trigger-Based Requalification Matrix
The table below defines typical trigger events and corresponding qualification expectations. Final scope must be determined through formal change control and risk assessment.
| Trigger Event | Impact Level | Typical Qualification Response |
|---|---|---|
| Minor maintenance with no flow path or parameter change | Low | Documented verification only |
| Sensor replacement (same specification and range) | Low–Moderate | Targeted OQ verification of affected function |
| Pump or valve replacement in product-contact path | Moderate | Partial IQ/OQ requalification of affected subsystem |
| Control system software update affecting recipes or alarms | Moderate–High | OQ re-execution including data integrity and alarm testing |
| Cleaning or sanitization chemistry modification | High | Cleaning revalidation and defined performance confirmation |
| Column type, resin, or pressure rating change | High | Full requalification including defined PQ execution |
| Adverse trend in pressure, capacity, or peak symmetry | Risk-based | Investigation; partial or full requalification depending on root cause |
9.2 Continued Verification
Requalification is event-driven. Continued verification is ongoing. Continued verification activities include:
• Trending backpressure to detect fouling or bed compression
• Monitoring dynamic binding capacity across cycles
• Evaluating peak symmetry and resolution
• Reviewing alarm frequency and interlock activations
• Assessing cleaning and sanitization performance consistency
Performance data should be reviewed periodically to confirm that the chromatography system remains within validated operating parameters. Trend instability, gradual degradation, or recurring deviations may trigger formal impact assessment and requalification.
9.3 Lifecycle Oversight
Effective lifecycle oversight integrates:
• Change control
• Periodic performance review
• Cleaning validation maintenance
• Calibration management
• Resin lifecycle monitoring
The objective is not repeated qualification, but sustained demonstration that the chromatography system remains fit for its intended purification function within a regulated environment.
10. Documentation Framework
Qualification documentation typically includes:
• User Requirement Specification
• Risk Assessment
• DQ, IQ, OQ, PQ protocols and reports
• Cleaning validation reports
• Calibration records
• Change control documentation
Traceability must link user requirements, qualification evidence, and operational monitoring.
11. Conclusion
Chromatography system qualification requires structured evaluation of mechanical integrity, fluid control, instrumentation, software, cleaning strategy, and lifecycle monitoring. A risk-based qualification approach ensures that the system consistently supports validated purification processes within a regulated environment.