FDA Inspection Readiness
FDA inspection readiness is the sustained ability of a regulated establishment to demonstrate that its operations comply with applicable statutory, regulatory, application, and procedural requirements. It depends on effective quality systems, controlled operations, reliable records, knowledgeable personnel, and timely correction of identified deficiencies.
Inspection readiness cannot be created immediately before an investigator arrives. A facility may organize an inspection support room, assign escorts, prepare document-retrieval procedures, and train personnel for inspection conduct, but these activities do not compensate for weak controls, incomplete investigations, unreliable data, or differences between written procedures and actual practices.
An inspection-ready organization maintains CGMP compliance during routine operation and can retrieve accurate evidence showing how its facilities, utilities, equipment, processes, computerized systems, laboratory operations, and quality systems remain in a state of control.

Regulatory Authority and Inspection Scope
FDA’s basic authority to inspect regulated establishments is established by section 704 of the Federal Food, Drug, and Cosmetic Act, codified at 21 U.S.C. §374. Section 704 authorizes duly designated FDA personnel to enter and inspect covered establishments at reasonable times, within reasonable limits, and in a reasonable manner.
For pharmaceutical manufacturers, investigators may evaluate compliance with:
- The Federal Food, Drug, and Cosmetic Act
- 21 CFR Parts 210 and 211
- 21 CFR Part 11, where applicable
- Product-specific regulations
- Commitments in approved applications
- Registered manufacturing and testing operations
- Established specifications and written procedures
- Other requirements applicable to the products and operations being inspected
Section 704 establishes inspection authority; the applicable regulations and approved applications establish many of the requirements evaluated during the inspection.
FDA inspections are regulatory examinations used to determine whether an establishment complies with applicable requirements. The scope and depth of an inspection depend on the inspection type, assigned program, products and operations, compliance history, identified risks, and findings developed during the inspection. Not every inspection covers every quality-system element to the same extent.
FDA’s Office of Inspections and Investigations leads the agency’s field inspection and related operational activities.
The statutory inspection provisions are available in 21 U.S.C. §374.
Types of FDA Inspections
Inspection-readiness priorities depend partly on why FDA is conducting the inspection.
Surveillance Inspections
Surveillance inspections evaluate ongoing compliance with applicable CGMP requirements. FDA uses risk-based factors to prioritize pharmaceutical manufacturing establishments for surveillance inspection.
A surveillance inspection may examine selected quality systems, manufacturing operations, laboratory controls, facilities, equipment, materials, records, and corrective actions. The scope can expand when investigators identify significant deficiencies or unresolved risks.
For-Cause Inspections
A for-cause inspection may be initiated in response to information suggesting a potential compliance or product-quality problem. Possible triggers include:
- Product complaints
- Adverse events or safety signals
- Recalls
- Field-alert reports
- Data-integrity concerns
- Whistleblower allegations
- Significant deviations
- Cross-contamination concerns
- Sterility failures
- Information obtained during another inspection
- Evidence of possible unapproved or noncompliant operations
A for-cause inspection is generally directed toward the issue that prompted FDA’s attention, but related findings may broaden its scope.
Preapproval and Pre-license Inspections
A preapproval inspection may be conducted to support FDA’s evaluation of a drug application. The inspection may assess whether:
- The establishment is ready for commercial manufacturing
- Submitted manufacturing and testing information is accurate
- The proposed process is adequately developed and controlled
- Facilities, utilities, and equipment are appropriately qualified
- Analytical procedures are suitable and transferable
- Data supporting the application are complete and reliable
- The establishment can manufacture the proposed product in accordance with the application and CGMP requirements
Prelicense inspections perform a comparable function for applicable biological products and licensed operations.
Follow-Up and Compliance Inspections
FDA may conduct a follow-up or compliance inspection to evaluate corrections made in response to previous observations, regulatory correspondence, enforcement action, or other identified deficiencies.
The company should be able to demonstrate that corrective actions were implemented, their scope was adequate, their effectiveness was evaluated, and the underlying causes were addressed rather than merely correcting individual examples.
Continuous Inspection Readiness
Continuous inspection readiness begins with routine compliance. It requires the establishment to operate every day in the same controlled manner it would expect to demonstrate during an FDA inspection.
Core elements include:
- Effective quality-unit oversight
- Approved and current procedures
- Qualified facilities, utilities, and equipment
- Validated manufacturing and cleaning processes
- Controlled computerized systems
- Suitable laboratory methods and instruments
- Trained and qualified personnel
- Complete and accurate records
- Timely deviation and failure investigations
- Scientifically supported corrective and preventive actions
- Effective change control
- Periodic review and data trending
- Management awareness of significant quality risks
A documented validation lifecycle supports readiness by connecting requirements, risk assessments, qualification, validation, routine monitoring, change control, periodic review, and retirement.
Readiness assessments should evaluate actual evidence rather than relying only on procedure availability. An approved procedure does not demonstrate compliance when records are incomplete or employees routinely follow a different practice.
Quality-System Oversight
The quality unit should maintain independent and effective oversight of CGMP operations. Inspection readiness requires the quality unit to understand current risks and to have access to reliable information concerning:
- Deviations and investigations
- Out-of-specification and out-of-trend results
- Complaints and recalls
- Batch disposition
- Process and environmental trends
- Equipment and utility performance
- Calibration and preventive maintenance
- Supplier performance
- Data-integrity concerns
- Change controls
- CAPA implementation and effectiveness
- Validation and requalification status
- Regulatory commitments
A recurring backlog does not become acceptable because records remain open in a tracking system. Overdue investigations, CAPAs, periodic reviews, calibrations, maintenance work, or validation activities should be evaluated for cumulative risk and potential effects on product quality.
Management review should identify unresolved risks, recurring failures, deteriorating trends, and resource limitations before they affect the establishment’s state of control.
Documentation and Data Readiness
FDA investigators use records to determine what occurred, whether approved procedures were followed, how decisions were made, and whether the quality system identified and corrected problems.
Records should be:
- Complete
- Accurate
- Legible
- Contemporaneous
- Traceable
- Reviewed and approved as required
- Protected from unauthorized alteration
- Retained for the applicable period
- Readily retrievable
- Consistent with related records and observed operations
FDA expects CGMP data to be reliable and accurate. A sound data-governance program should ensure that data are attributable, legible, contemporaneously recorded, original or appropriately preserved, accurate, complete, consistent, enduring, and available throughout the required retention period.
These expectations are addressed further in ALCOA principles and data integrity and electronic-record lifecycle and retention.
Records Commonly Reviewed
Depending on the inspection scope, FDA may request:
- Organization charts and facility information
- Site quality manuals
- Validation Master Plans
- Product and process lists
- Batch production and control records
- Specifications and analytical procedures
- Qualification and validation protocols and reports
- Cleaning-validation records
- Environmental-monitoring data
- Utility monitoring and trending
- Calibration and maintenance records
- Deviations and investigations
- Out-of-specification investigations
- Complaints and recalls
- CAPA records
- Change controls
- Training records
- Supplier qualification records
- Computerized-system validation records
- Audit trails and data reviews
- Annual product reviews or product-quality reviews
- Management-review records
A controlled validation-documentation structure should establish traceability between requirements, risk assessments, protocols, executed results, deviations, reports, approvals, and lifecycle records.
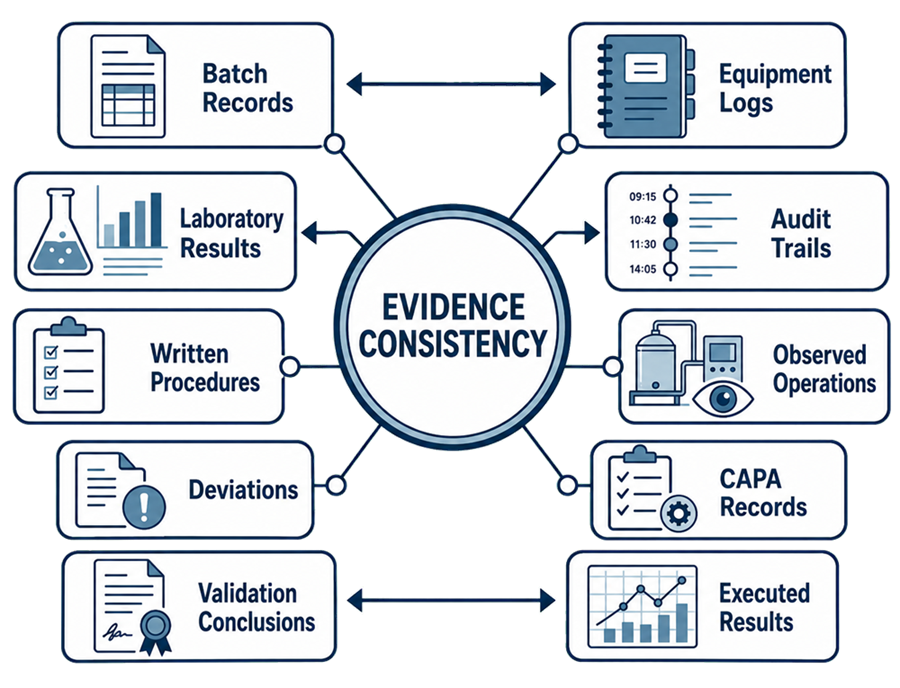
Record Consistency
Investigators may compare different sources of evidence, such as:
- Batch records against equipment-use logs
- Laboratory results against audit trails
- Procedures against observed operator practices
- Training records against assigned responsibilities
- Deviations against CAPA records
- Change controls against configuration history
- Calibration records against equipment use
- Environmental data against investigation decisions
- Validation conclusions against executed results
- Application commitments against current manufacturing practices
Contradictory records, unexplained data gaps, reconstructed documentation, and differences between procedures and actual practices can raise broader questions about the quality system.
Data Integrity and Computerized Systems
Computerized systems used for CGMP operations should be validated and controlled according to their intended use and risk.
Inspection readiness for computerized systems should address:
- Authorized user access
- Role assignments and segregation of duties
- Unique user identification
- Audit-trail generation and review
- Electronic signatures
- Configuration control
- Data acquisition and processing
- Calculations and reports
- Interfaces and data transfers
- Backup and restoration
- Record retention and retrieval
- System security
- Incident management
- Change control
- Periodic review
- System retirement and data migration
The company should be able to explain how original electronic data, metadata, audit trails, calculations, reprocessing activities, and result changes are reviewed.
Testing only printed reports may be insufficient when the electronic system contains relevant underlying data or metadata. Audit-trail review requirements should be based on the record’s CGMP significance, the system’s functionality, and the opportunity for changes affecting reported results.
Relevant controls are addressed in computerized-system audit trails, access control and electronic signatures, and backup, restoration, and recovery.
Facility and Operational Readiness
The physical condition of the facility should agree with approved drawings, procedures, qualification records, and stated contamination-control practices.
Routine readiness reviews should examine:
- Material and personnel flows
- Facility zoning and segregation
- Cleaning and sanitation
- Equipment identification and status
- Equipment condition
- Calibration status
- Preventive-maintenance status
- Logbook completion
- Line clearance
- Material labeling and status control
- Storage conditions
- Pest-control evidence
- Waste handling
- Prevention of contamination and mix-ups
- Environmental-monitoring practices
- Utility-system operation
- Access restrictions
- Temporary repairs and inactive equipment
Facility conditions that have become normalized internally may be readily apparent during an independent walkthrough. Examples include damaged finishes, unidentified equipment, accumulated materials, uncontrolled tools, incomplete logbooks, ineffective segregation, obsolete labels, temporary tubing arrangements, or undocumented repairs.
A strong calibration and maintenance control program helps demonstrate that critical equipment remains capable of performing its intended function.
Personnel Interviews and Inspection Conduct
Personnel should understand their assigned responsibilities and be able to explain how they perform their work. Inspection preparation should reinforce accurate communication, not scripted responses.
Employees should:
- Listen carefully to the question
- Answer truthfully and directly
- Provide the information needed for an accurate answer
- Describe actual approved practices
- Avoid speculation or guessing
- Ask for clarification when the question is unclear
- Identify the appropriate subject-matter expert when necessary
- State when information must be verified from a record
- Avoid making unsupported commitments
- Remain professional throughout the interaction
A subject-matter expert (SME) is the individual with sufficient knowledge and responsibility to explain a particular system, process, record, or technical decision.
Personnel should not conceal relevant facts, provide knowingly incomplete answers, alter records, or attempt to coordinate misleading explanations. Verification is appropriate when information is not known or cannot be confirmed from memory.
Differences between employee explanations, written procedures, records, and observed practices can expand the inspection scope and raise questions about the effectiveness of training and quality oversight.
Preparing for an FDA Inspection
An inspection-management procedure should define responsibilities before FDA arrives. The procedure should address:
- Reception and notification
- Verification of investigator credentials
- Senior-management and quality-unit notification
- Legal or regulatory notification, where appropriate
- Facility access
- Safety and security requirements
- Opening-meeting responsibilities
- Investigator escorts
- Record requests
- Document review and reproduction
- Electronic-record access
- Facility photography or recording requests
- Sample collection
- Daily inspection summaries
- Observation tracking
- Closeout preparation
- Post-inspection response
The company should designate primary and alternate personnel for critical roles. The process must remain functional if an inspection begins when specific senior employees are unavailable.
Inspection Support Room
An inspection support room can coordinate:
- Investigator requests
- Document retrieval
- SME availability
- Document-production status
- Copies provided to FDA
- Questions requiring follow-up
- Potential observations
- Daily management updates
The support room should facilitate accurate and timely responses. It should not be used to alter responsive records, conceal information, coordinate misleading answers, or delay production without legitimate reason.
Opening the Inspection
At the beginning of an inspection, FDA personnel generally present their credentials and may issue Form FDA 482, Notice of Inspection, when applicable.
The opening meeting should establish:
- Investigator identities
- Inspection purpose and expected scope
- Facility contacts
- Safety requirements
- Working arrangements
- Record-request procedures
- Electronic-system access arrangements
- Daily schedules
- Communication expectations
Company representatives should record the investigator’s requests and clarify ambiguous requests promptly. They should not misrepresent facility operations or unnecessarily restrict access authorized by law.
The inspection scope can change as the investigator reviews records, observes operations, interviews personnel, and develops findings.
Managing FDA Record Requests
Each request should be recorded using a controlled tracking process. The request log should capture:
- Request number
- Exact request
- Date and time received
- Responsible owner
- Status
- Clarifications
- Documents or information provided
- Date and time completed
- Follow-up requests
Requested records should be checked for:
- Completeness
- Legibility
- Correct revision
- Accurate identification
- Responsiveness to the request
- Inclusion of required attachments
- Consistency with related submissions
The company should maintain a copy of each record provided to FDA and document any explanations accompanying it.
The company should not alter, reconstruct, conceal, or improperly withhold responsive records. Questions concerning request scope, privileged material, personal information, confidential commercial information, or other legally protected material should be escalated immediately to designated quality, regulatory, and legal representatives.
When a requested record cannot be located promptly, the company should communicate its status accurately and continue a documented search. Creating a replacement record or backdating missing documentation is unacceptable.
Electronic Records and System Access
FDA may request access to electronic records and the systems used to create, modify, process, review, approve, or retain them.
The company should be prepared to:
- Identify the authoritative electronic record
- Explain system functionality
- Demonstrate user roles and permissions
- Retrieve records for the requested period
- Display relevant metadata and audit trails
- Explain calculation or processing steps
- Demonstrate backup and restoration controls
- Identify original and reprocessed data
- Explain data review and approval
- Provide controlled electronic or printed copies
Temporary accounts created for inspection purposes should have documented, appropriate permissions. Granting unrestricted access without considering system security and record protection may create additional risk.
Applicable requirements for electronic records and signatures are addressed in 21 CFR Part 11 compliance.
Advance Record Requests and Remote Regulatory Assessments
FDA may request records before or in advance of an inspection under applicable authority. The same controls used for on-site document production should apply to advance submissions.
The establishment should:
- Assign responsibility for each request
- Confirm the requested scope
- Track submission deadlines
- Verify record completeness
- Maintain copies of submitted materials
- Use secure and controlled transmission methods
- Document explanations and follow-up responses
- Escalate potential compliance concerns identified during retrieval
FDA may also conduct a Remote Regulatory Assessment. An RRA is an examination of an FDA-regulated establishment and its records conducted entirely remotely. An RRA is not itself an inspection under sections 704(a)(1) or 704(a)(5) of the FD&C Act.
A remote assessment should nevertheless be managed through controlled record retrieval, verified submissions, qualified SMEs, secure communication, accurate representations, and documented follow-up.
FDA explains these mechanisms in its Conducting Remote Regulatory Assessments guidance.
Daily Inspection Management
The inspection team should review activities at the end of each inspection day. The review should identify:
- Requests received
- Responses completed
- Outstanding records
- Follow-up questions
- Areas visited
- Personnel interviewed
- Samples collected
- Potential observations
- Factual misunderstandings
- Immediate corrections
- Emerging product or compliance risks
Potential deficiencies should be evaluated promptly. Immediate correction may be appropriate, but correction during the inspection does not eliminate the need to determine root cause, assess scope, evaluate product impact, implement sustainable corrective action, and document effectiveness.
The company should avoid arguing about defensible factual points during routine interactions. Material factual errors should be clarified using accurate records and qualified personnel.
Inspection Closeout
At the conclusion of an inspection, FDA generally conducts a closeout discussion with senior management. If investigators have identified conditions that, in their judgment, may constitute violations, they may issue Form FDA 483.
The closeout meeting allows management to:
- Understand each observation
- Request clarification
- Identify material factual errors
- Provide relevant information not previously considered
- Describe corrections already completed
- Explain planned corrective actions
- Confirm future communication arrangements
Management should avoid making unsupported commitments or promising completion dates before the required scope, resources, and technical work have been evaluated.
A company should document the closeout discussion, including verbal comments that may not appear on Form FDA 483.
Form FDA 483
FDA may issue Form FDA 483 to firm management at the conclusion of an inspection when investigators observe conditions that, in their judgment, may constitute violations of the FD&C Act or related requirements.
Form FDA 483 communicates inspectional observations. It is not an all-inclusive list of every possible deficiency, a final agency determination, or a final enforcement action.
FDA considers the Form FDA 483 together with:
- The Establishment Inspection Report
- Evidence and records collected during the inspection
- The company’s written response
- Corrections and supporting documentation
- Compliance history
- Product and patient risk
- Other relevant information
FDA discusses the observations with senior management during the inspection closeout. The company should ensure that it understands the scope and factual basis of each observation before preparing its response.
FDA provides additional explanation in its Form FDA 483 Frequently Asked Questions.
Responding to Form FDA 483
A Form FDA 483 response should be specific, evidence-based, and proportionate to the risk and systemic significance of the observations. It should not consist primarily of promises, revised procedures, or employee retraining without adequate investigation.
For each observation, the response should address:
- Acknowledgment and the company’s position
- Immediate containment
- Corrections already completed
- Product-quality and patient-risk assessment
- Root-cause investigation
- Retrospective scope assessment
- Corrective and preventive actions
- Interim controls
- Responsible owners
- Realistic completion dates
- Supporting documentation
- Effectiveness checks
- Follow-up commitments for incomplete actions
Product and Scope Assessment
The response should determine whether the observation affects:
- Distributed or released product
- Other batches
- Other products
- Other equipment or systems
- Other departments
- Other sites
- Submitted regulatory data
- Ongoing stability studies
- Pending applications
- Previously closed investigations or CAPAs
Correcting the individual example cited by FDA is insufficient when the underlying cause may affect a wider population.
Root Cause and CAPA
Root-cause conclusions should be supported by evidence. A conclusion such as operator error, inadequate training, or failure to follow procedure should not be used without determining why the existing system allowed the event to occur and why it was not detected earlier.
CAPA should address the underlying control failure and include an appropriate method for verifying effectiveness.
Revised procedures and retraining may be components of CAPA, but they do not automatically demonstrate that the process, system, management controls, resources, or oversight deficiencies have been corrected.
Response Timing
FDA generally encourages firms to submit a written response within 15 business days so that it can be considered during the agency’s review. When all corrective actions cannot be completed within that period, the response should explain:
- Work already completed
- Remaining actions
- Interim risk controls
- Justified completion dates
- The schedule for providing supporting evidence
Completion dates should reflect the actual work required. Unsupported or repeatedly missed commitments can undermine the credibility of the response.
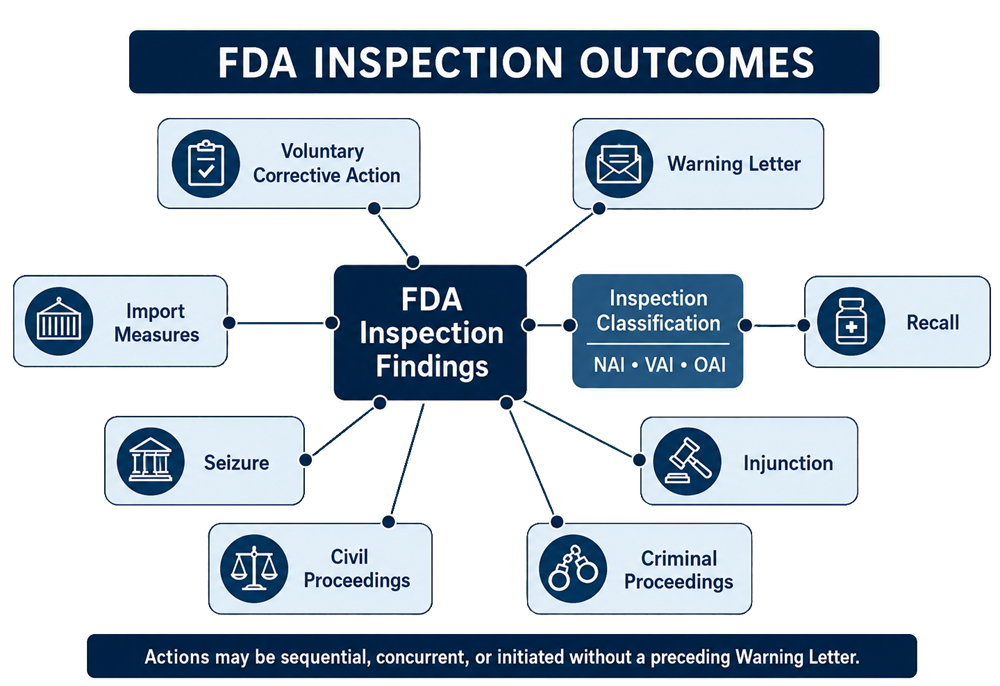
Inspection Classifications
After reviewing the inspection record and related evidence, FDA may classify the inspection as NAI, VAI, or OAI.
No Action Indicated — NAI
NAI generally indicates that no objectionable conditions or practices warranting action were found during the inspection.
An NAI classification does not establish that every operation was examined or that the establishment is permanently compliant. The company remains responsible for maintaining compliance and correcting internally identified deficiencies.
Voluntary Action Indicated — VAI
VAI indicates that objectionable conditions or practices were found, but FDA is not prepared to take or recommend regulatory or administrative action. The establishment is expected to correct the deficiencies voluntarily.
A VAI classification should not be interpreted as acceptance of the observed conditions or as relief from the responsibility to implement effective corrective action.
Official Action Indicated — OAI
OAI indicates that regulatory or administrative action is recommended based on the inspection findings and the agency’s evaluation.
The specific outcome depends on the violations, product and patient risk, compliance history, corrective response, legal authority, and other case-specific considerations.
FDA describes these classifications in its Inspection Classification Database.
Regulatory and Enforcement Outcomes
FDA responses to inspection findings do not follow a mandatory escalation ladder. Depending on the facts and applicable authority, actions may be sequential, concurrent, or initiated without a preceding Warning Letter.
Potential outcomes include:
- No further regulatory action
- Voluntary corrective action
- Regulatory meetings
- Warning Letter
- Import measures
- Recall
- Seizure
- Injunction
- Consent decree
- Civil proceedings
- Criminal investigation or prosecution
The appropriate response depends on the regulatory significance of the violations, product and patient risk, compliance history, recurrence, adequacy of corrective actions, management response, evidence of misconduct, and applicable legal authority.
Warning Letters
The U.S. Food and Drug Administration may issue a Warning Letter when documented violations are of regulatory significance and prompt, adequate correction is expected.
A Warning Letter is an official advisory action used to obtain voluntary compliance and establish prior notice. It is not a final agency action.
A Warning Letter generally communicates:
- The violations identified by FDA
- The regulatory significance of the findings
- FDA’s requested corrective response
- The response timeframe
- Possible consequences of inadequate correction
FDA considers case-specific factors such as violation severity, product risk, compliance history, recurrence, previous notice, corrective actions, and management response.
A Warning Letter is not a prerequisite to seizure, injunction, prosecution, import action, or another enforcement measure. FDA’s Regulatory Procedures Manual states that enforcement strategies may be sequential or concurrent.
Import Measures
For products offered for import into the United States, FDA may use import controls when applicable legal criteria are met. An import alert may allow detention without physical examination of covered products.
Import measures can affect products, establishments, manufacturers, or geographic areas depending on the basis and scope of the alert. Removal from an import alert generally requires evidence addressing the underlying compliance concerns.
Recalls
A recall is generally an action taken by a firm on its own initiative or at FDA’s request to remove or correct a marketed product that presents a regulatory or health concern. Specific mandatory-recall authorities exist for certain product categories and circumstances, but drug recalls should not be described universally as compulsory FDA enforcement actions.
An effective recall system should provide:
- Accurate product and lot traceability
- Rapid distribution identification
- Defined recall responsibilities
- Health-hazard evaluation support
- Effectiveness checks
- Reconciliation
- Regulatory communication
- Disposition of returned product
Seizure
Seizure is a judicial action against violative products. It may prevent the distribution or continued availability of affected products while the legal proceeding is resolved.
Injunction and Consent Decree
An injunction is a court order restraining continued violations or imposing corrective conditions.
A consent decree generally results from a negotiated resolution of a federal injunction proceeding and becomes enforceable through a court order. Its provisions may restrict operations until specified corrective conditions are satisfied and may remain effective for an extended period.
Consent decrees can result in substantial financial and operational costs and may impose:
- Long-term remediation requirements
- Independent expert oversight
- Periodic reporting
- FDA review and inspection
- Restrictions on manufacturing or distribution
- Product recalls or destruction
- Payments associated with regulatory oversight
- Conditions for resuming operations
Criminal Investigations
When evidence indicates possible criminal violations, the matter may be referred to the FDA Office of Criminal Investigations (OCI) for evaluation and, where appropriate, criminal investigation in coordination with prosecuting authorities.
Criminal investigations may involve:
- Fraud
- Intentional falsification
- Obstruction
- Counterfeiting
- Diversion
- Deliberate concealment
- Tampering
- Illegal distribution
- Other suspected criminal conduct
Whether a matter warrants criminal investigation depends on the evidence and applicable law. FDA describes OCI’s responsibilities in What We Investigate.
FDA’s procedures for Warning Letters and other regulatory actions are described in the Regulatory Procedures Manual.
Internal Readiness Assessments
An internal inspection-readiness assessment should test whether the establishment can demonstrate compliance using actual records, employee explanations, facility observations, and electronic data.
The assessment may include:
- Review of previous FDA observations and commitments
- Review of regulatory correspondence
- Evaluation of recurring deviations
- CAPA effectiveness review
- Data-integrity review
- Facility walkthroughs
- Batch-record review
- Laboratory-data review
- Validation-status review
- Utility and environmental-data review
- Computerized-system review
- Employee interviews
- Record-retrieval exercises
- Mock request management
- Evaluation of inspection procedures
The assessment should prioritize significant product-quality and data-reliability risks. It should not focus primarily on cosmetic documentation changes or rehearsed responses while systemic problems remain unresolved.
Findings should enter the established quality system and be corrected through documented risk assessment, investigation, CAPA, change control, or other appropriate processes.
Common Inspection-Readiness Weaknesses
Inspection-readiness programs commonly fail when:
- Readiness activity begins only after inspection notice
- Procedures do not reflect actual practice
- Deviations are closed without adequate root-cause analysis
- CAPAs correct examples without addressing systemic scope
- Recurring events are investigated independently
- Validation conclusions are unsupported by executed data
- Equipment is used with overdue calibration or maintenance
- Computerized systems lack adequate access or audit-trail controls
- Records cannot be retrieved promptly
- Electronic and paper records conflict
- Employees provide speculative or inconsistent explanations
- Management lacks awareness of significant quality risks
- Commitments are made without realistic plans or resources
- Internal audit findings remain open or repeatedly recur
A documented change-control impact assessment and effective periodic review help detect cumulative changes and degradation before they become inspection findings.
Bottom Line
FDA inspection readiness is the ability to demonstrate sustained compliance through controlled operations, reliable data, complete records, knowledgeable personnel, and effective quality oversight.
Section 704 of the FD&C Act establishes FDA’s basic inspection authority. The requirements evaluated during a pharmaceutical inspection may arise from Parts 210, 211, and 11; approved applications; product-specific regulations; established specifications; written procedures; and other applicable commitments.
Inspection management can organize communication, personnel, records, and logistics, but it cannot create compliance. The strongest readiness program is a functioning pharmaceutical quality system that identifies risks, investigates failures, implements effective corrective action, and maintains facilities, systems, and processes in a state of control.
Form FDA 483 is not a final agency determination, and regulatory outcomes do not follow a mandatory escalation sequence. A defensible response must address product impact, root cause, systemic scope, CAPA, supporting evidence, effectiveness, responsible owners, and realistic completion dates.