Validation V-Model in GMP and Regulated Manufacturing
Validation within pharmaceutical, biotechnology, medical device, and other FDA-regulated manufacturing environments requires a structured lifecycle approach that demonstrates systems are designed, installed, tested, operated, and maintained in a controlled and traceable manner. Validation activities provide documented evidence that facilities, utilities, equipment, processes, and computerized systems consistently perform according to predefined requirements, regulatory expectations, and intended use.
The validation V-model, also referred to as the verification and validation model, provides a systematic framework that links system requirements and design activities with corresponding qualification and verification activities. The model is widely applied throughout regulated industries because it supports traceability, quality risk management, lifecycle control, and maintenance of the validated state throughout the operational life of the system.
Overview of the V-Model
The V-model is represented graphically as the letter “V”:
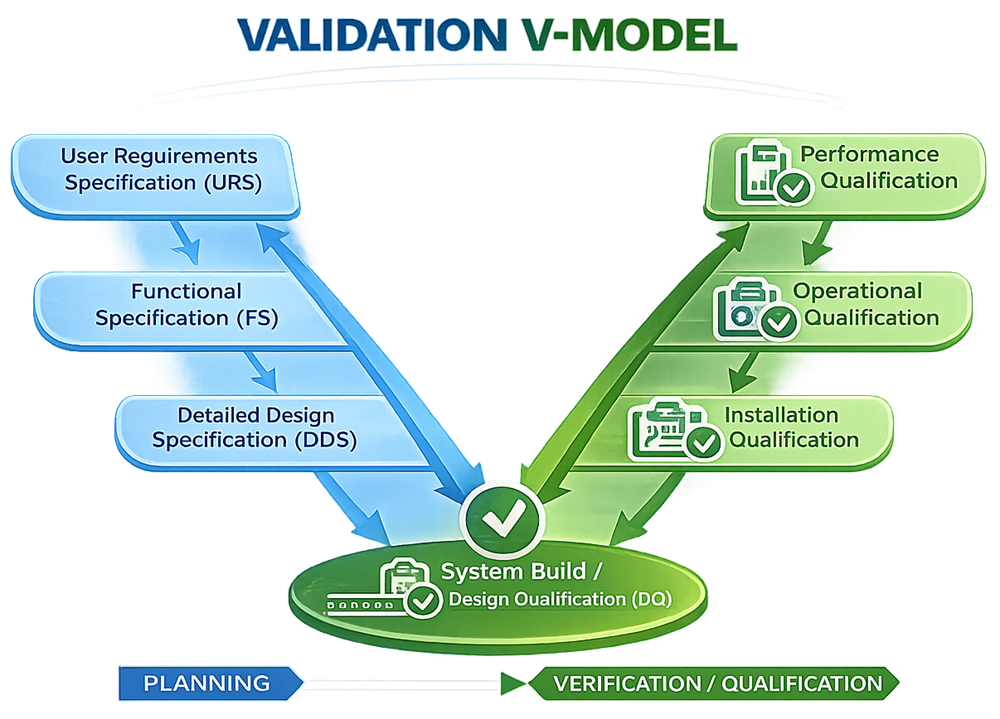
The left side of the model represents planning, specification, and design activities where system requirements and technical definitions are developed. The bottom portion of the model represents system implementation and installation.
The right side of the model represents qualification, verification, and testing activities where documented evidence is generated to confirm that the system performs according to approved requirements and specifications.
A core principle of the V-model is that each specification or design activity has a corresponding verification or qualification activity. This structure establishes traceability between user requirements, design documentation, testing activities, and final system acceptance.
The V-model is commonly applied to manufacturing equipment, process systems, utility systems, cleanroom automation, computerized systems, laboratory instruments, packaging systems, and sterile manufacturing operations.
User Requirements Specification (URS)
The User Requirements Specification (URS) defines what the system must accomplish from the user, operational, business, and regulatory perspectives. The URS establishes high-level requirements related to functionality, capacity, performance, compliance, alarms, security, data integrity, environmental conditions, and operational expectations.
The URS serves as the foundation for system design, procurement, risk assessment, testing strategy, and qualification activities. Well-developed URS documentation helps ensure that validation activities remain focused on critical business and GMP requirements rather than unnecessary testing activities.
All downstream specifications, design documents, and qualification protocols should remain aligned with the approved URS.
Functional Design Specification (FDS)
The Functional Design Specification (FDS) describes how the system will satisfy the requirements defined within the URS. The FDS translates user and regulatory requirements into functional descriptions, operating logic, system behaviors, process flows, alarms, interfaces, and control strategies.
The FDS focuses on intended system functionality without yet defining detailed implementation-level construction or configuration details. This document establishes the functional basis for software development, automation configuration, system integration, and operational testing.
The FDS should remain traceable to the URS to demonstrate that all critical requirements have been addressed within the proposed design.
Detailed Design Specification (DDS)
The Detailed Design Specification (DDS) defines how the system will be physically and technically implemented. Depending on system complexity, the DDS may include engineering drawings, electrical schematics, network architecture, software configuration details, control logic, component specifications, hardware layouts, piping diagrams, instrumentation details, and configuration parameters.
The DDS serves as the technical blueprint for construction, installation, configuration, and implementation activities. The document should remain aligned with both the URS and FDS to maintain traceability throughout the validation lifecycle.
For computerized systems and automation platforms, the DDS may also define security configuration, backup strategies, audit trail functionality, user access management, and data integrity controls.
Design Qualification (DQ)
Design Qualification (DQ) provides documented verification that the proposed system design is suitable for its intended use and capable of meeting user, operational, engineering, and regulatory requirements.
DQ activities typically involve documented review of the URS, FDS, DDS, vendor documentation, risk assessments, material specifications, and supporting engineering documents. The objective is to confirm that the proposed design is technically appropriate before fabrication, procurement, installation, or configuration activities begin.
Effective DQ activities help identify design deficiencies early in the project lifecycle when corrective actions are less costly and easier to implement.
Installation Qualification (IQ)
Installation Qualification (IQ) verifies and documents that the system has been installed according to approved design specifications, manufacturer recommendations, engineering documentation, and applicable standards. IQ activities commonly include verification of:
- Equipment identification
- Component installation
- Utilities and connections
- Materials of construction
- Instrumentation
- Calibration status
- Software installation
- Environmental conditions
- Network configuration
- Documentation completeness
Supporting records such as calibration certificates, as-built drawings, manuals, wiring diagrams, spare parts lists, and vendor documentation are typically reviewed during IQ execution. Successful IQ establishes the documented foundation required for operational testing activities.
Operational Qualification (OQ)
Operational Qualification (OQ) demonstrates that the system operates according to approved functional specifications throughout defined operating ranges and anticipated conditions. OQ testing focuses on:
- Functional operation
- Alarm testing
- Interlocks
- Control sequences
- Security functions
- Software functionality
- Automation logic
- Data collection
- User access controls
- System responses under normal and abnormal conditions
Acceptance criteria should be predefined and scientifically justified based on system requirements, risk assessment results, and intended use. OQ provides documented evidence that the system functions correctly prior to routine operational use.
Performance Qualification (PQ)
Performance Qualification (PQ) demonstrates that the system consistently performs effectively under actual or simulated routine operating conditions.
PQ activities verify that the system supports intended operational processes, manufacturing activities, and user requirements over time using representative materials, loads, recipes, products, or operating conditions where appropriate. Depending on system type and criticality, PQ activities may include:
- Production simulations
- Process runs
- Environmental monitoring
- Extended operation
- User workflows
- Batch execution activities
- Data review
- Process consistency evaluation
Successful PQ establishes documented evidence that the system is suitable for ongoing GMP use within the regulated manufacturing environment.
Traceability Within the Validation V-Model
One of the most important strengths of the validation V-model is traceability between requirements, specifications, testing activities, and final system acceptance.
Traceability matrices are commonly used to demonstrate that:
- User requirements defined within the URS are addressed within the FDS and DDS
- Design specifications are verified during qualification testing
- Critical requirements are tested and documented
- Deviations and discrepancies are evaluated appropriately
- Final acceptance is supported by objective evidence
This traceability structure supports inspection readiness, change control assessment, deviation investigations, lifecycle management, and long-term maintenance of the validated state.
Validation Lifecycle and Ongoing Control
The validation V-model should not be viewed as a one-time project activity. Validation continues throughout the operational lifecycle of the system through activities such as:
- Change control
- Calibration
- Preventive maintenance
- Backup verification
- Periodic review
- Continued process verification
- Environmental monitoring
- Deviation management
- Incident investigation
- Requalification activities
Maintaining the validated state requires continued oversight to ensure that systems remain controlled, compliant, reliable, and suitable for their intended use as operating conditions, processes, software, and regulatory expectations evolve over time.
Conclusion
The validation V-model provides a structured and traceable lifecycle framework that links system requirements, design activities, qualification testing, and operational verification within GMP-regulated environments.
By establishing clear relationships between specifications and qualification activities, the V-model supports regulatory compliance, quality risk management, inspection readiness, and long-term maintenance of the validated state across pharmaceutical, biotechnology, medical device, laboratory, and other regulated manufacturing systems.